Huntington’s Disease Symptoms: Huntington’s Disease (HD) is a progressive and fatal genetic disorder that affects the brain, leading to severe physical and mental symptoms.
Characterized by its autosomal dominant inheritance pattern, this means that a child has a 50% chance of inheriting the condition if one parent is affected.
Understanding the symptoms and causes of Huntington’s Disease is crucial for early diagnosis and management.
What is Huntington’s Disease?
Huntington’s disease (HD) is a progressive brain disorder caused by a defective gene. This disease gradually breaks down nerve cells in the brain, impacting physical movements, cognitive abilities, and emotions. Symptoms of Huntington’s disease commonly appear between the ages of 30 and 50, and the condition typically progresses over a 10 to 25-year period. Ultimately, the disorder causes a wide range of symptoms, including motor control loss, cognitive decline, and psychiatric issues.
Genetic Basis of the Disease
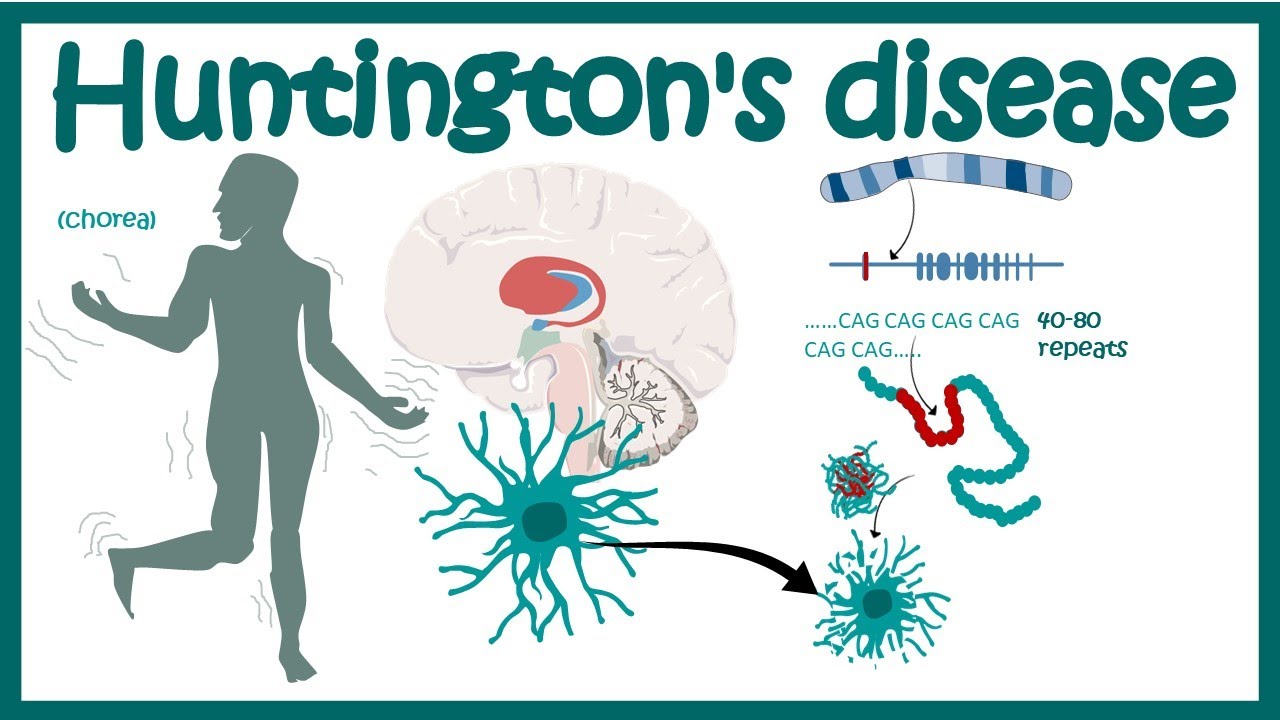
The genetic cause of Huntington’s disease lies in the mutation of the HTT gene, which provides instructions for making a protein called huntingtin. The mutation involves an abnormal expansion of a DNA segment in this gene. This expansion leads to the production of an abnormal version of the huntingtin protein, which accumulates and causes damage within the brain’s nerve cells. Everyone has two copies of the HTT gene, but only one mutated gene is sufficient to cause the disorder. This is known as an autosomal dominant pattern of inheritance.
Prevalence and Demographic Data
Huntington’s disease affects about 5 to 7 per 100,000 people of European ancestry. The disease is less prevalent in other populations, such as those of Japanese, Chinese, and African descent, where the prevalence is below one per 100,000. The condition affects males and females equally and typically manifests in adulthood, although there is a less common juvenile form that begins in childhood or adolescence. Despite its rarity, the impact of Huntington’s disease is significant, affecting numerous families across generations.
Symptoms of Huntington’s Disease
Here, we’ll explore the early signs and symptoms of Huntington’s Disease, understand how these symptoms progress over time, and examine their impact on daily life and independence.
Early Signs and Symptoms of Huntington’s Disease
The initial symptoms of Huntington’s Disease can vary widely among individuals but commonly include subtle changes in coordination and mood. Key early symptoms include:
- Mood Swings and Depression: Individuals may experience frequent mood swings, irritability, or depression.
- Subtle Coordination Problems: Early signs might include slight uncontrolled movements, clumsiness, or balance issues.
- Cognitive Changes: There might be difficulties with problem-solving, decision-making, or focusing on tasks.
- Memory Lapses: Forgetting names, appointments, or recent events can occur in the early stages.
Recognizing these signs can be crucial for early diagnosis and management of the disease.
Progression of Symptoms Over Time
As Huntington’s Disease progresses, symptoms become more pronounced and debilitating:
- Motor Skills Deterioration: Jerky, random, and uncontrollable movements known as chorea increase and intensify.
- Cognitive Decline: Cognitive abilities gradually worsen, affecting the ability to think clearly and perform complex tasks.
- Psychological Impact: Emotional disturbances, including increased depression, anxiety, and sometimes psychotic episodes, become more common.
- Communication Difficulties: Speech becomes slurred and hard to understand, and understanding others can also be challenging.
The progression rate can vary, but these symptoms typically increase in severity and significantly impact daily functioning.
Impact on Daily Life and Independence
The symptoms of Huntington’s Disease profoundly affect an individual’s daily life and independence:
- Daily Activities and Self-Care: Simple tasks such as dressing, eating, and personal hygiene become challenging, often requiring assistance from caregivers.
- Mobility Issues: Increased difficulty with physical coordination can lead to frequent falls and the need for mobility aids.
- Social Interaction: Communication challenges and cognitive decline make social interactions difficult, potentially leading to social withdrawal.
- Emotional Toll: The psychological impact of managing a chronic, progressive disease can be significant, affecting both the individual and their family.
Living with Huntington’s Disease requires adapting to these changes, and support from healthcare providers, therapists, and support groups is crucial.
However, as symptoms evolve, they increasingly impact daily activities and independence, highlighting the need for comprehensive care strategies and support systems to maintain quality of life.
Causes of Huntington’s Disease
Understanding its causes is crucial for those at risk and for medical professionals managing the condition. Below, we delve into the genetic origins and inheritance patterns of Huntington’s disease, explain the HTT gene mutation, and list risk factors that may influence the development of HD.
Genetic Origins and Inheritance Patterns
Huntington’s disease is primarily caused by genetic mutations and follows an autosomal dominant inheritance pattern. This means a single copy of the altered gene in each cell is sufficient to cause the disorder. The gene responsible for HD is known as the HTT gene. If one parent has a defective HTT gene, each child has a 50% chance of inheriting the gene and, consequently, the disease. This pattern of inheritance holds consistent across both genders and all ethnic groups.
Explanation of the HTT Gene Mutation
The HTT gene mutation responsible for Huntington’s disease involves the expansion of a DNA segment known as a CAG trinucleotide repeat. In individuals without HD, the CAG segment is typically repeated 10 to 35 times within the gene. However, in individuals with HD, this segment is repeated 36 to more than 120 times. The severity and onset of the disease are often related to the number of repeats: the greater the number of repeats, the earlier the onset and the more severe the symptoms. This mutation results in the production of an abnormal version of the huntingtin protein, which gradually damages certain brain cells, leading to the symptoms of HD.
Risk Factors for Developing Huntington’s Disease
The primary risk factor for Huntington’s disease is having a parent with the condition due to its autosomal dominant genetic transmission. However, other factors can influence the onset and progression of the disease:
- Number of CAG Repeats: As previously mentioned, a higher number of CAG repeats in the HTT gene typically correlates with earlier onset and more rapid progression of the disease.
- Age of Onset in Parents: The age at which a parent began showing symptoms can influence the age of onset in the offspring, with younger parental onset sometimes leading to earlier and more severe manifestations in the next generation.
- Environmental Factors: While genetic factors are predominant, environmental factors and overall health can influence disease progression and symptom severity.
However, understanding these factors is vital for genetic counseling and for managing expectations and preparations for those at risk of Huntington’s disease.
Diagnosing Huntington’s Disease
Early and accurate diagnosis is crucial for managing symptoms and planning future care. Below, we discuss common diagnostic tests and procedures that help in identifying HD, with a focus on genetic testing and counseling.
Common Diagnostic Tests and Procedures
- Neurological Examination: This is usually the first step in diagnosing Huntington’s Disease. During a neurological exam, a doctor will assess the patient’s motor, sensory, and mental functions. The examination includes testing reflexes, muscle strength, muscle tone, sense of touch, and balance.
- Psychiatric Evaluation: Since HD can affect mental health, a psychiatric assessment is essential. This evaluation helps in identifying any emotional and behavioral changes, which are common in patients with HD. Psychiatrists may assess mood, mental state, and behavior as part of the diagnosis.
- Brain Imaging and Scans: Imaging tests such as MRI (Magnetic Resonance Imaging) and CT (Computed Tomography) scans are used to detect changes in the brain’s structure that are indicative of Huntington’s Disease. These scans can help in assessing the extent of brain degeneration and ruling out other conditions.
- Neuropsychological Testing: These tests assess cognitive functions that may be affected by HD, such as memory, problem-solving skills, and the ability to plan and organize. Neuropsychological testing can help in determining the impact of the disease on cognitive abilities.
Genetic Testing and Counseling
Genetic testing is a definitive method for diagnosing Huntington’s Disease. Since HD is caused by a specific genetic mutation, identifying this mutation through genetic testing confirms the diagnosis.
- Procedure of Genetic Testing: The test involves a simple blood draw from which DNA is extracted. The laboratory then examines the DNA for the specific genetic mutation associated with HD, namely the HTT gene mutation. This test can also be used predictively to assess the risk before symptoms appear, especially in individuals with a family history of the disease.
- Genetic Counseling: Because of the hereditary nature of HD and the implications of genetic testing, genetic counseling is recommended both before and after the test. Genetic counselors provide crucial support and information, helping individuals and families understand the results and implications of the test. They discuss potential outcomes, reproductive options, and psychological impacts of knowing one’s genetic status.
However, engaging with healthcare providers to determine the best approach based on individual and family circumstances is recommended for anyone concerned about Huntington’s Disease.
Impact of Symptoms on Daily Life of Huntington’s Disease
Huntington’s disease profoundly impacts the daily lives of those affected, influencing physical abilities, emotional well-being, and social interactions. Understanding these impacts can provide valuable insights into the challenges faced by individuals and their families.
Physical Impact
- Motor Function Decline: Huntington’s disease commonly causes involuntary movements, muscle rigidity, and poor coordination. These symptoms can significantly impair the ability to perform routine tasks such as walking, driving, or handling utensils.
- Difficulty with Speech and Swallowing: Progressive loss of muscle control affects speech and swallowing, complicating communication and eating, which can lead to nutritional deficiencies and weight loss.
- General Physical Decline: As the disease progresses, overall physical health deteriorates, leading to increased dependency on caregivers for daily activities and personal care.
Emotional and Mental Health Challenges
- Cognitive Decline: Individuals often experience a decline in cognitive functions, including memory, planning, and decision-making, which can contribute to feelings of frustration and loss of independence.
- Mood Disorders: Depression and irritability are common in people with Huntington’s disease, stemming from both neurological changes and the psychological stress of coping with a chronic illness.
- Anxiety: The uncertainty about disease progression and the impact on life expectancy can lead to significant anxiety, affecting mental health and overall quality of life.
Social and Familial Effects
- Isolation: Physical symptoms and emotional changes can make social interactions challenging, leading to withdrawal and isolation from friends and community activities.
- Family Relationships: The hereditary nature of Huntington’s disease can place a strain on family relationships, with members facing fears about their own health and potential caregiving responsibilities.
- Caregiver Burden: Families often bear a heavy burden in providing care, which can impact their emotional and financial stability, affecting family dynamics and relationships.
However, addressing these challenges requires a comprehensive support system involving medical treatment, emotional counseling, and community support to help improve the quality of life for those affected by Huntington’s disease.
Treatment and Management of Huntington’s Disease
Various treatment options are available to help manage symptoms and improve the quality of life for those affected. This article outlines the current treatment options, explores ongoing research into new treatments, and provides tips for lifestyle adjustments and home care.
Current Treatment Options
1. Medication: Various medications can help manage the symptoms of HD. These include:
- Antipsychotics (such as haloperidol and risperidone) to help control emotional and behavioral problems.
- Antidepressants (like sertraline and fluoxetine) to tackle mood swings and depression.
- Tetrabenazine and deutetrabenazine are specifically approved to treat chorea associated with Huntington’s disease, helping to reduce involuntary movements.
2. Physical Therapy: Regular physical therapy can assist in maintaining mobility and balance, which can prevent falls and help individuals remain independent for longer.
3. Speech Therapy: As HD progresses, speech therapy can be beneficial in helping patients maintain their communication skills and manage swallowing difficulties.
Research on New Treatments
Scientists are continuously researching new treatments to slow the progression of Huntington’s Disease and alleviate symptoms more effectively. Some promising areas of research include:
- Gene Therapy: Efforts are being made to develop therapies that target the genetic root of the disease, potentially slowing or halting its progression.
- Stem Cell Therapy: Researchers are exploring the use of stem cells to regenerate damaged brain cells and restore neurological function.
- Novel Medications: Clinical trials are ongoing for new drugs that aim to improve the neurological function or target the underlying disease mechanisms in HD.
Lifestyle Adjustments and Home Care Tips
Living with Huntington’s Disease requires adjustments to daily life and routines. Here are some tips to make home care more effective:
- Adapt Your Home: Make modifications such as installing grab bars and removing loose rugs to create a safer environment.
- Routine Establishment: Keeping a consistent daily routine can help manage cognitive challenges like memory loss or confusion.
- Nutritional Support: Swallowing difficulties are common, so it’s essential to ensure the diet is easy to consume and nutritionally balanced.
- Emotional and Social Support: Regular interaction with friends, family, or support groups can provide emotional stability and decrease feelings of isolation.
However, consulting healthcare providers for personalized advice and staying informed about new research can significantly aid in navigating this challenging condition.
Prevention and Genetic Counseling of Huntington’s Disease
Huntington’s Disease (HD) is a hereditary neurological disorder with significant implications for affected families. While there is currently no cure for HD, prevention through genetic counseling plays a crucial role in managing the disease’s impact on future generations.
Role of Genetic Counseling in Prevention
Genetic counseling for Huntington’s Disease serves as a vital preventive tool by offering at-risk individuals and families informed choices about their health and reproductive decisions. Genetic counselors provide essential services that include:
- Risk Assessment: Counselors evaluate the likelihood of an individual inheriting or passing on HD by analyzing family history and, if applicable, conducting genetic tests.
- Education about HD: Individuals receive comprehensive information about the nature of the disease, its symptoms, progression, and the implications of genetic testing.
- Support in Decision Making: Genetic counselors assist individuals and families in understanding their reproductive options, including prenatal testing and in-vitro fertilization with preimplantation genetic diagnosis (PGD), which can prevent the transmission of the disease to offspring.
- Emotional and Psychological Support: Dealing with the risk or reality of HD can be distressing. Counselors provide support and can refer individuals to appropriate resources, including support groups and psychological services.
Information for At-Risk Individuals
For individuals at risk of Huntington’s Disease, obtaining accurate and comprehensive information is critical. Genetic counseling offers a confidential environment where individuals can explore their concerns about the disease. Information provided includes:
- Genetic Testing Process: Details on how genetic testing is done, its benefits, limitations, and the implications of knowing one’s genetic status.
- Management of HD: While prevention of the disease itself is not currently possible, counselors can provide information on managing symptoms and improving quality of life for those diagnosed with HD.
- Legal and Ethical Considerations: Counselors discuss the potential impacts of genetic information on insurance, employment, and personal relationships.
However, genetic counseling is indispensable for at-risk individuals, offering them vital information and support that aids in making informed decisions about their health and family planning.
FAQs about Huntington’s Disease Symptoms
What are the early symptoms of Huntington’s Disease?
Early symptoms of Huntington’s Disease often include subtle changes in mood, cognition, and motor skills. Common early signs are slight uncontrolled movements, difficulty in thinking through problems, and occasional mood swings. These symptoms may initially be mistaken for stress or aging.
How do the symptoms of Huntington’s Disease progress?
As Huntington’s Disease progresses, symptoms become more pronounced and interfere significantly with daily activities. Motor symptoms may evolve into chorea, which involves involuntary, jerky movements. Cognitive decline also progresses, leading to difficulties with memory, planning, and decision-making. Emotional symptoms may include depression, irritability, and apathy.
Can the symptoms of Huntington’s Disease vary between individuals?
Yes, symptoms of Huntington’s Disease can vary widely between individuals. The age of onset and the progression of the disease can differ significantly. Some may experience more severe motor symptoms early on, while others might have more pronounced cognitive or emotional issues.
Are there any late-stage symptoms of Huntington’s Disease that caregivers should be aware of?
In the late stages of Huntington’s Disease, individuals may experience severe motor impairment, requiring assistance with all daily activities. Cognitive abilities continue to decline, often resulting in dementia. Emotional disturbances might also intensify. Caregivers should be prepared for comprehensive care needs and consider seeking support from health services specialized in Huntington’s Disease.
Is it possible to predict the symptoms and progression of Huntington’s Disease?
Predicting the exact symptoms and progression of Huntington’s Disease in an individual can be challenging. Genetic testing can determine if a person will develop the disease, which often helps in anticipating some aspects of its progression. However, the specific symptoms and their severity cannot be precisely predicted and vary from person to person.
Conclusion
In summary, Huntington’s disease presents with a range of challenging symptoms, including cognitive decline, motor dysfunctions, and emotional disturbances. Understanding these symptoms is crucial for those affected by the disease and their families.
We encourage everyone impacted by Huntington’s to continue educating themselves and seeking out robust support networks. Knowledge and community support can significantly enhance the quality of life for those dealing with this condition.
Stay informed, connected, and proactive in managing the challenges of Huntington’s disease.
References
For further reading and validation of the information provided on Huntington’s Disease symptoms, we recommend consulting the following reputable sources:
- Mayo Clinic – Offers comprehensive information on the symptoms, causes, and treatment options for Huntington’s Disease. Read more here.
- National Institute of Neurological Disorders and Stroke (NINDS) – Provides detailed insights into the neurological aspects of Huntington’s Disease, including current research and advancements. Learn more here.
- Huntington’s Disease Society of America (HDSA) – A valuable resource for understanding the genetic and community aspects of Huntington’s Disease, with access to support networks and advocacy information. Explore more here.
- MedlinePlus – A trusted resource for medical information, MedlinePlus provides an easy-to-understand overview of Huntington’s Disease symptoms and management. Check it out here.
These sources offer reliable and in-depth information, ensuring you have access to the most accurate and up-to-date knowledge on Huntington’s Disease.


