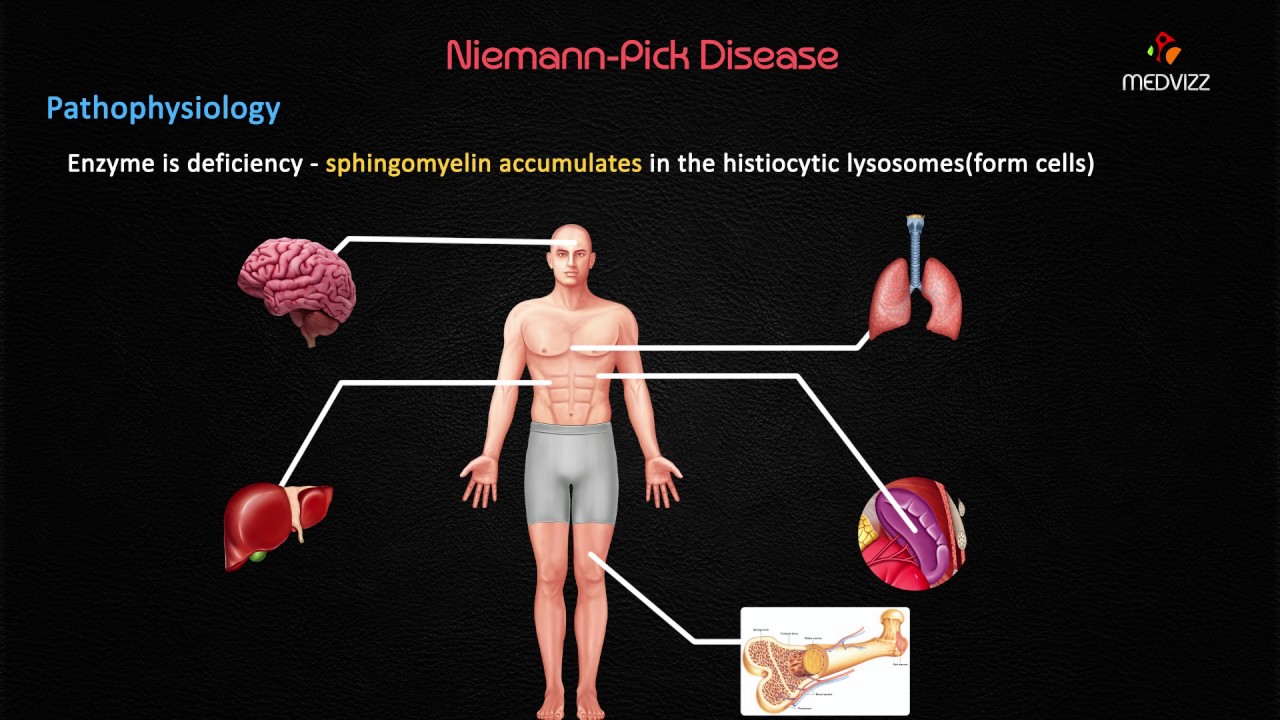
Niemann-Pick Disease Symptoms: Niemann-Pick disease is a rare, inherited disorder that affects the body’s ability to metabolize fat (lipids) within cells.
These cells, which become dysfunctional due to the accumulation of fats, are found primarily in the liver, spleen, bone marrow, and brain.
This accumulation can cause a variety of health problems, ranging from mild to severe.
What is Niemann-Pick Disease?
Niemann-Pick disease is a rare, inherited condition that involves the metabolism of lipids in the body. This disruption leads to the accumulation of harmful quantities of lipids in various organs, including the brain, liver, and spleen. The disease primarily affects children, but symptoms and onset can vary widely depending on the type of Niemann-Pick disease involved. Common symptoms include an enlarged liver and spleen, neurological impairment, and significant developmental delays. Due to its genetic nature, Niemann-Pick disease is caused by mutations in specific genes that are responsible for lipid metabolism.
Types of Niemann-Pick Disease
Niemann-Pick disease is categorized into several types, each distinguished by their genetic cause and associated symptoms:
- Type A: This form is severe, typically manifesting in early infancy. Children with Type A experience rapid neurological degeneration, enlarged liver and spleen, and rarely live beyond their early childhood years.
- Type B: Unlike Type A, Type B usually presents later in childhood or adolescence and primarily affects the spleen and liver. Neurological involvement is less severe or absent, allowing individuals with Type B to survive into adulthood.
- Type C: Type C can appear in childhood or as late as adulthood, characterized by its neurological symptoms such as difficulty moving and speaking, seizures, and progressive loss of motor skills. This type is not related to the enzyme deficiency found in Types A and B but rather to a defect in cholesterol transport.
- Type D: Often grouped with Type C, Type D is similar in symptoms but has a specific genetic mutation found principally in individuals of Nova Scotian descent.
However, each type of Niemann-Pick disease presents unique challenges in diagnosis and management, making awareness and early detection critical for improving outcomes.
Common Symptoms of Niemann-Pick Disease
Understanding the common symptoms, how they differ between types, and their effects on quality of life is crucial for early diagnosis and management.
Early Symptoms and Signs
The early signs of Niemann-Pick disease can vary but generally include:
- Enlarged liver and spleen (hepatosplenomegaly): Often the first noticeable symptom, which can occur in infancy or early childhood.
- Failure to thrive: Infants may not gain weight or grow at the expected rate.
- Difficulty feeding: This includes challenges with sucking or swallowing.
- Developmental delays: Delays in reaching milestones such as sitting, standing, or walking.
- Frequent respiratory infections.
As the disease progresses, symptoms may become more severe and include neurological impairment such as loss of muscle tone and coordination, learning difficulties, and seizures.
Variations in Symptoms Between Types
Niemann-Pick disease is categorized mainly into three types: A, B, and C, each differing significantly in symptoms:
- Type A: This is the most severe form, usually manifesting in early infancy. Symptoms include rapid neurodegeneration, severe liver dysfunction, and is typically fatal by early childhood.
- Type B: Although similar liver and spleen enlargement are observed, individuals with Type B usually do not experience the neurological symptoms seen in Type A and can live into adulthood.
- Type C: Symptoms may not appear until childhood or adolescence and include neurological symptoms like ataxia, vertical supranuclear gaze palsy, and dementia. Unlike Types A and B, Type C involves impaired movement of cholesterol and other fats.
Impact on Quality of Life
The quality of life for individuals with Niemann-Pick disease can be profoundly affected. The symptoms can lead to:
- Physical limitations: Difficulty in normal physical activities due to enlarged organs, poor muscle tone, and neurological degeneration.
- Cognitive decline: Especially in Types A and C, where neurological involvement can lead to a decline in mental functioning, affecting learning abilities and daily living skills.
- Social challenges: The visible symptoms and physical limitations can lead to social isolation and emotional challenges for patients and their families.
- Life expectancy: Particularly in Type A and severe cases of Type C, the life expectancy can be significantly shortened.
Management of Niemann-Pick disease focuses on symptom relief and supportive care, as there is no cure currently available. Early intervention and supportive therapies can help manage the symptoms and improve the quality of life for those affected by the disease.
Causes of Niemann-Pick Disease
Niemann-Pick disease is a rare genetic disorder primarily caused by mutations in specific genes that affect lipid metabolism. This results in the harmful accumulation of lipids in various body tissues, including the brain, liver, and spleen. The disease manifests in different forms, each with distinct genetic origins and inheritance patterns.
Genetic Origins and Inheritance Patterns
- Type A and B: Both Type A and B Niemann-Pick disease are caused by mutations in the SMPD1 gene. This gene is responsible for producing the enzyme acid sphingomyelinase (ASM). Deficiency in ASM leads to the accumulation of sphingomyelin, a type of lipid, in cells. The inheritance pattern of these types is autosomal recessive, meaning that an individual must inherit two copies of the mutated gene, one from each parent, to be affected.
- Type C: Niemann-Pick Type C disease is predominantly caused by mutations in either the NPC1 or NPC2 genes. These genes are crucial for the movement of cholesterol and other lipids within cells. Mutations in these genes disrupt this process, leading to lipid accumulation. Like Types A and B, Type C follows an autosomal recessive inheritance pattern.
Differences in Cause Between Type A, B, and C
- Type A: This form is characterized by a severe deficiency of the ASM enzyme. The onset is typically in infancy, and the disease progression is rapid and severe, leading to early childhood death.
- Type B: Although also resulting from mutations in the SMPD1 gene, Type B is less severe. Patients have a partial deficiency of the ASM enzyme. Symptoms are more varied, and individuals with Type B often survive into adulthood.
- Type C: The cause of Type C is distinct from Types A and B because it involves different genes (NPC1 or NPC2) and affects cholesterol trafficking rather than sphingomyelinase activity. The onset can occur anytime from early childhood to adulthood, with symptoms and severity varying widely among individuals.
However, understanding these genetic and biochemical distinctions is crucial for diagnosing the specific type of Niemann-Pick disease and for tailoring appropriate treatment and management strategies.
Diagnosing Niemann-Pick Disease
Here we explore the common diagnostic tests and procedures, as well as the role of genetic testing in accurately identifying this condition.
Common Diagnostic Tests and Procedures
- Physical Examination: Initially, healthcare providers conduct a thorough physical examination, looking for typical signs such as an enlarged liver or spleen, which are characteristic of Niemann-Pick Disease.
- Blood Tests: Blood tests help in assessing liver function and cholesterol levels, both of which can be affected by Niemann-Pick Disease. These tests can indicate abnormal functioning that may suggest the presence of a storage disease.
- Imaging Studies: Techniques like ultrasound, MRI (Magnetic Resonance Imaging), or CT (Computed Tomography) scans are employed to visualize internal organs. For Niemann-Pick Disease, these imaging tools can show enlargement of the spleen and liver.
- Biopsy: A definitive diagnostic tool for Niemann-Pick Disease is the biopsy, often of the liver or bone marrow. In this procedure, a small tissue sample is taken and examined for the abnormal storage of materials, which is a hallmark of the disease.
- Enzyme Assay: This test measures the activity of specific enzymes in white blood cells or fibroblasts cultured from a skin biopsy. A deficiency in certain enzymes, such as acid sphingomyelinase in Niemann-Pick types A and B, confirms the diagnosis.
Role of Genetic Testing in Diagnosing Niemann-Pick Disease
Genetic testing plays a pivotal role in diagnosing Niemann-Pick Disease, particularly for confirming the type and informing treatment strategies:
- Identifying Mutations: Genetic tests can identify mutations in the SMPD1 gene responsible for Types A and B, or the NPC1 and NPC2 genes associated with Type C. Detecting these mutations provides a clear diagnosis, distinguishing among the disease types, which is essential for determining the appropriate management approach.
- Family Screening: Once a mutation is identified in an individual, genetic testing can be used to screen family members. This is crucial for understanding the inheritance pattern and providing early interventions for affected relatives.
- Guiding Treatment: The results from genetic testing can guide treatment decisions. For instance, certain experimental therapies may be more applicable to one type of Niemann-Pick Disease than another, depending on the underlying genetic cause.
However, each tool provides valuable information that contributes to a comprehensive understanding of the disease, aiding in the effective management and care of affected individuals.
Treatment Options for Niemann-Pick Disease
Managing this condition involves a combination of current treatments and promising research that points toward future therapies.
Current Treatments Available
1. Enzyme Replacement Therapy (ERT): For types of Niemann-Pick disease that result from enzyme deficiencies, ERT can be effective. This treatment involves administering the deficient enzyme to patients, helping to break down the accumulated fats in cells.
2. Symptomatic Management: Due to the diverse symptoms of Niemann-Pick, a multidisciplinary approach is necessary. This includes:
- Physical therapy to enhance mobility and reduce the risk of muscle atrophy.
- Nutritional support to address feeding difficulties and ensure proper nutrition.
- Medication to manage symptoms such as seizures, sleep disturbances, and lung infections.
3. Bone Marrow Transplantation: In some cases, particularly in children with severe manifestations, bone marrow transplantation has been used to introduce healthy cells that can produce the enzyme that is deficient.
4. Supportive Care: Regular monitoring by healthcare professionals including neurologists, pulmonologists, and gastroenterologists to manage the systemic effects of the disease.
Research and Future Potential Treatments
1. Gene Therapy: Researchers are exploring gene therapy as a potential cure for Niemann-Pick disease. This treatment involves correcting the genetic mutations that cause the disease, aiming to enable the body to produce the missing enzyme or protein naturally.
2. Small Molecule Drugs: These drugs aim to assist in the proper folding and functioning of proteins or enzymes, potentially reducing the accumulation of fats in cells. This approach is still in the early stages of research.
3. Substrate Reduction Therapy (SRT): SRT reduces the substrate (the substance upon which an enzyme acts), thereby decreasing the amount of lipid accumulation in cells. This method is showing promise in clinical trials.
4. Chaperone Therapy: This treatment uses chaperone molecules to stabilize enzymes and ensure they function correctly. It is currently under investigation and may provide a viable treatment option in the future.
However, ongoing research continues to push the boundaries of what is possible, aiming to improve quality of life and longevity for patients with this challenging condition.
Living with Niemann-Pick Disease
Living with Niemann-Pick Disease (NPD) presents unique challenges and requires careful management to improve quality of life and mitigate symptoms. NPD is a group of inherited metabolic disorders where harmful quantities of lipids accumulate in various organs, including the brain. While treatment options can vary depending on the type and severity of the disease, there are several strategies and resources that can help manage symptoms and provide support for individuals and families affected by NPD.
Management Strategies for Symptoms
- Medication: Specific medications may be prescribed to help manage symptoms or slow the progression of the disease. For example, enzyme replacement therapies are being explored for certain types of NPD.
- Nutritional Support: A dietitian may recommend specific dietary adjustments that help manage symptoms, such as a low-cholesterol diet or nutritional supplements to support overall health.
- Physical Therapy: Regular physical therapy can help maintain mobility and function, reducing the physical complications associated with muscle weakness and coordination issues.
- Occupational Therapy: This can assist individuals in maintaining their independence by developing strategies to manage daily activities and improve quality of life.
- Speech Therapy: Especially useful for children, speech therapy can help manage difficulties with speech that arise from the disease’s impact on the muscles involved in speaking.
- Regular Monitoring and Check-ups: Regular visits with healthcare providers are crucial for monitoring the progression of the disease and adjusting treatment plans as needed.
Support Resources: Organizations and Support Groups
- National Niemann-Pick Disease Foundation (NNPDF): Offers a wide range of resources including educational materials, support groups, and connections to clinical trials.
- International Niemann-Pick Disease Alliance (INPDA): A global network that connects various national groups providing support and information sharing among those affected by NPD.
- The Global Genes Project: Focuses on connecting and supporting individuals with rare diseases, including NPD. They offer tools, educational resources, and events.
- RareConnect: Includes online communities for patients and families to connect with others worldwide facing similar rare diseases, facilitating the exchange of information and experiences.
- Local Hospitals and Clinics: Often host support groups for patients and families dealing with genetic disorders, providing a local and immediate network of support.
- Social Media Platforms: Various groups and pages dedicated to NPD can be found on platforms like Facebook, where members share advice, experiences, and emotional support.
By leveraging these management strategies and resources, individuals with Niemann-Pick Disease and their families can find meaningful support and effective ways to cope with the challenges posed by the condition.
FAQs about Niemann-Pick Disease Symptoms
What are the primary symptoms of Niemann-Pick disease?
Niemann-Pick disease symptoms vary significantly between types A, B, and C. Common symptoms across all types include an enlarged liver and spleen, difficulty in movement, and issues with growth. Type A often presents severe neurological symptoms early in life, such as loss of motor skills and feeding difficulties. Type B usually involves respiratory problems but less severe neurological symptoms. Type C primarily causes neurological symptoms such as tremors, difficulty swallowing, and cognitive decline.
How early do symptoms of Niemann-Pick disease appear?
Symptoms of Niemann-Pick disease can appear at different times for each type. Type A symptoms typically manifest in early infancy, often within a few months after birth. Type B symptoms might appear in late childhood or adolescence, while Type C can vary widely, showing up in childhood or even as late as adulthood.
Can Niemann-Pick disease symptoms improve over time?
Currently, there is no cure for Niemann-Pick disease, and symptoms generally progress over time. However, treatments can help manage symptoms and improve quality of life. For example, enzyme replacement therapy has shown promise in managing some symptoms of Type B. Supportive treatments for Type C may include medications to manage seizures and psychiatric symptoms.
Are there any specific triggers that worsen symptoms of Niemann-Pick disease?
Triggers can vary, but in general, infections and illnesses may exacerbate symptoms due to the added stress on the body’s systems. For individuals with Type C, certain medications can interfere with already compromised neurological functions and should be used cautiously under medical supervision.
Is it possible to mistake Niemann-Pick disease for another condition?
Yes, due to its varied symptoms and the rarity of the disease, Niemann-Pick can often be mistaken for other disorders, especially in its early stages. It shares symptoms with other lysosomal storage diseases, liver diseases, and neurological conditions. Accurate diagnosis typically requires genetic testing and lipid profiling to confirm the presence of specific biomarkers associated with Niemann-Pick disease.
Conclusion
In conclusion, recognizing the symptoms and understanding the causes of Niemann-Pick Disease is crucial for early diagnosis and effective management. This rare, inherited condition can significantly impact patients’ quality of life, making awareness and knowledge key to improving outcomes.
We encourage further research into this disease to uncover new treatments and interventions. Additionally, increasing public awareness can foster a supportive community for those affected.
By continuing to educate ourselves and support research, we can make significant strides in combating the challenges posed by Niemann-Pick Disease.
References
For further reading and to validate the information provided on the treatment of Dyshidrosis, consider the following reputable sources. These references offer comprehensive insights and are instrumental in understanding the various aspects of managing this skin condition:
- National Eczema Association – Provides detailed guidance on managing Dyshidrosis, including treatment options and lifestyle recommendations. Available at: National Eczema Association
- DermNet NZ – Offers an extensive overview of Dyshidrosis, covering symptoms, causes, and treatment modalities, supported by dermatological expertise. Visit: DermNet NZ Dyshidrosis Page
- American Academy of Dermatology – Features practical advice and treatment strategies from leading dermatologists for those suffering from Dyshidrosis. Explore more at: American Academy of Dermatology
These sources are valuable for both patients and healthcare providers seeking to deepen their understanding of Dyshidrosis and its treatment options.


