Creutzfeldt-Jakob Disease Symptoms: Creutzfeldt-Jakob Disease (CJD) is a rare, degenerative, invariably fatal brain disorder that significantly impacts about one in every one million people worldwide annually.
This condition belongs to a family of human and animal diseases known as the transmissible spongiform encephalopathies (TSEs) or prion diseases.
The onset of symptoms typically occurs at around age 60, and the average duration of illness is about a year.
What is Creutzfeldt-Jakob Disease?
Creutzfeldt-Jakob Disease (CJD) is a rare, degenerative, invariably fatal brain disorder, affecting about one in every one million people worldwide annually. Characterized by rapid neurological deterioration, usually leading to death within a year of onset, CJD belongs to a family of diseases known as prion diseases or transmissible spongiform encephalopathies (TSEs). These diseases affect both humans and animals, causing the brain tissue to become abnormally shaped and leading to significant impairment in brain function.
Brief History and Discovery
The disease was first described in the 1920s by two German neurologists, Hans Gerhard Creutzfeldt and Alfons Maria Jakob, hence the name Creutzfeldt-Jakob Disease. Their documentation of the disease laid the groundwork for understanding a range of neurodegenerative conditions that were later discovered to be caused by prions. Prions are misfolded proteins that replicate by converting their properly folded counterparts into the misfolded structure, leading to brain damage and the characteristic symptoms of CJD and related diseases.
Types of Creutzfeldt-Jakob disease
CJD can be categorized into several types, each with distinct causes and characteristics:
- Sporadic CJD (sCJD): The most common form, accounting for approximately 85% of cases. It occurs spontaneously, without known cause, in individuals with no risk factors.
- Hereditary CJD (familial CJD or fCJD): Accounts for about 10-15% of cases. It is caused by a genetic mutation and can run in families. Those with a family history of the disease are at increased risk.
- Variant CJD (vCJD): Linked to the consumption of meat from cattle affected by Bovine Spongiform Encephalopathy (BSE), also known as “mad cow disease.” vCJD is much rarer and tends to affect younger individuals compared to sCJD.
- Iatrogenic CJD: Results from medical procedures, including the transplantation of infected tissue or the use of contaminated surgical instruments. It is extremely rare, thanks to improved sterilization and screening practices.
Each type of CJD has its own onset, progression, and epidemiology, but they all share the common feature of rapid cognitive decline, motor dysfunction, and ultimately, death. Understanding these types is crucial for diagnosis, management, and prevention strategies.
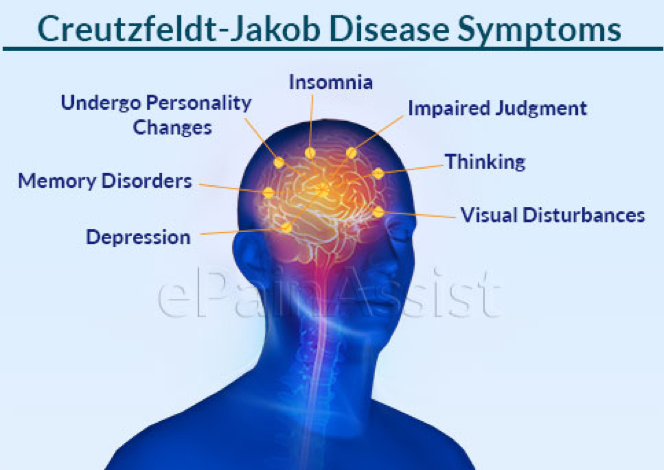
Symptoms of Creutzfeldt-Jakob Disease
Symptoms of CJD can be categorized into three stages: early, progressive, and late-stage. Each stage is marked by distinct symptoms, reflecting the progressive nature of the disease.
Early Symptoms
The early symptoms of CJD are often subtle and can easily be mistaken for signs of other, less severe neurological conditions. These initial symptoms include:
- Memory loss: Patients may experience forgetfulness or minor memory lapses.
- Behavioral changes: Slight changes in personality or mood swings may be noticeable.
- Visual disturbances: Some individuals may report difficulties with vision, such as blurriness or blind spots.
- Lack of coordination: Difficulty walking or clumsy movements can occur.
- Fatigue: A general feeling of tiredness or exhaustion without a clear cause.
Progressive Symptoms
As CJD progresses, the symptoms become more pronounced and debilitating. During the progressive stage, individuals may experience:
- Increased memory loss and confusion: Memory problems become more severe, leading to significant confusion.
- Advanced coordination problems: Movements become more uncoordinated, making walking or simple tasks challenging.
- Muscular stiffness: Patients may experience muscle stiffness or spasms.
- Speaking difficulties: Speech may become slurred or hard to understand.
- Severe visual impairment: Vision problems may worsen, potentially leading to blindness.
Late-stage Symptoms
In the late stages of CJD, symptoms become severe, and patients require comprehensive care:
- Complete immobility: The individual may become bedridden due to loss of muscle control.
- Mute: The ability to speak is lost.
- Incontinence: Loss of bladder and bowel control.
- Swallowing difficulties: This can lead to a risk of choking or aspiration pneumonia.
- Cognitive impairment: Severe dementia is common, with the patient losing the ability to recognize family or surroundings.
Timeline of Symptom Progression
The progression of symptoms in CJD can be rapid. From the onset of the first symptoms, the disease typically progresses quickly:
- Early symptoms can manifest subtly and may be overlooked initially.
- Progressive symptoms tend to develop within weeks to a few months following the onset of early symptoms.
- Late-stage symptoms often occur within a year of diagnosis, though the timeline can vary from person to person.
The rapid progression of CJD highlights the importance of seeking medical attention at the first sign of any neurological symptoms. Early diagnosis and supportive care are vital in managing the symptoms and improving the quality of life for those affected by this devastating disease.
Causes of Creutzfeldt-Jakob Disease (CJD)
Understanding the causes of CJD is crucial for prevention and management of the disease. This condition is attributed to abnormal forms of prion proteins that damage brain cells. Below, we delve into the roles of prion proteins and the different causes of CJD, including sporadic, hereditary, and acquired forms.
The Role of Prion Proteins
Prion proteins are naturally occurring and usually harmless. However, when they fold into abnormal shapes, they can become pathogenic, leading to diseases such as CJD. These misfolded proteins can accumulate in the brain, causing cell death, brain damage, and the characteristic symptoms of CJD. The exact mechanism of how normal prion proteins turn into their disease-causing form remains a subject of research.
Sporadic CJD Causes
Sporadic CJD (sCJD) is the most common form of the disease, accounting for about 85% of cases. It occurs randomly, with no identifiable cause. The hypothesis is that it might result from the spontaneous misfolding of normal prion proteins into the abnormal form. Why this happens remains largely unknown, making sporadic CJD a perplexing condition for scientists and medical professionals.
Hereditary CJD Causes
Approximately 10-15% of CJD cases are hereditary, caused by a genetic mutation in the prion protein gene (PRNP). Individuals who inherit the mutated gene from a parent have a higher risk of developing CJD. Testing for this genetic mutation can help identify at-risk individuals, although the presence of the mutation does not guarantee disease development.
Genetic Mutations
The specific mutations in the PRNP gene vary among families and can influence the age of onset and symptoms of the disease. Research is ongoing to understand how these genetic variations affect the progression and characteristics of CJD.
Acquired CJD Causes
Acquired CJD is the least common form and results from external sources of infection. Two primary ways through which CJD can be acquired include:
- Medical Procedures: In rare cases, CJD has been transmitted through medical procedures that involved the use of contaminated instruments or tissues, such as brain surgery with inadequately sterilized equipment or transplantation of infected tissues.
- Consumption of Contaminated Meat (Variant CJD): Variant CJD (vCJD) is linked to consuming meat from cattle affected by bovine spongiform encephalopathy (BSE), also known as mad cow disease. This form of CJD has a younger age of onset compared to other types and has been primarily identified in the UK and other countries affected by BSE outbreaks.
While the sporadic form remains a mystery, awareness and control measures can significantly reduce the risk of hereditary and acquired CJD. Ongoing research into prion diseases may one day provide insights into their prevention, diagnosis, and treatment, offering hope for those affected by this devastating condition.
Risk Factors and Transmission of Creutzfeldt-Jakob Disease (CJD)
Understanding the risk factors and transmission modes of CJD is crucial for prevention and management of this disease. This article will explore genetic predisposition, exposure risks, and the rarity of transmission, providing valuable insights into the complexities of CJD.
Genetic Predisposition
One of the primary risk factors for developing CJD is genetic predisposition. Approximately 10-15% of CJD cases are familial, which means they are caused by inherited mutations in the prion protein gene. Individuals with a family history of CJD are at a higher risk of developing the disease. Genetic testing can identify mutations in the prion protein gene, offering crucial information for families with a history of CJD. Understanding your genetic risk can aid in early diagnosis and management, although it is important to consult with healthcare professionals and genetic counselors for personalized advice.
Exposure Risks
Exposure to brain or nervous system tissue from an infected individual or animal can also pose a risk for transmitting CJD. This can occur through certain medical procedures, such as:
- Surgical Instruments: Although extremely rare, there is a possibility of CJD transmission through surgical instruments previously used on an infected patient if the instruments were not properly sterilized.
- Corneal Transplants: There have been reported cases of CJD transmission through corneal transplants from donors with undiagnosed CJD.
- Human Growth Hormone (hGH) Treatments: Prior to 1985, some patients who received hGH derived from human pituitary glands developed CJD. Since then, synthetic hGH has eliminated this risk.
It is essential for healthcare facilities to follow stringent sterilization and screening protocols to minimize these risks.
Understanding the Rarity of Transmission
The transmission of CJD is exceedingly rare, and the disease is not contagious through casual contact, such as touching or being near someone with CJD. There is no evidence to suggest that CJD can be spread through air, water, soil, or consuming meat from animals other than the specific variant known as variant Creutzfeldt-Jakob Disease (vCJD), linked to bovine spongiform encephalopathy (BSE) in cattle.
Understanding the rarity of transmission is important to alleviate unwarranted fears and stigma associated with the disease. Education and awareness about the actual risks and modes of transmission can help in preventing unnecessary alarm and ensuring that those affected by CJD receive the support and care they need.
However, while genetic predisposition and certain types of exposure can increase the risk of developing CJD, it is important to remember that the overall risk remains very low. Advances in medical practices, including the use of synthetic hormones and improved sterilization techniques, have further reduced the risks of transmission. Public health measures and ongoing research continue to play a vital role in managing and understanding CJD.
Diagnosis and Detection of Creutzfeldt-Jakob Disease
Early detection and accurate diagnosis are crucial for managing this condition, as they can help in planning treatment options and supportive care. This article explores the diagnosis and detection of CJD, emphasizing the significance of medical history, symptoms, diagnostic tests, and the importance of early detection.
Medical History and Symptoms
The initial step in diagnosing CJD involves a thorough review of the patient’s medical history and a detailed assessment of symptoms. Early symptoms of CJD can include memory problems, behavioral changes, visual disturbances, and poor coordination. As the disease progresses, mental deterioration becomes pronounced, leading to muscle stiffness, twitching, and weakness. A comprehensive evaluation of these symptoms, in conjunction with a patient’s medical history, can guide healthcare professionals towards suspecting CJD.
Diagnostic Tests
Several diagnostic tests are instrumental in confirming CJD:
- Magnetic Resonance Imaging (MRI): MRI scans are crucial for detecting changes in brain structure caused by CJD. They can reveal specific patterns of brain atrophy and abnormalities that are indicative of the disease.
- Spinal Fluid Tests: The analysis of cerebrospinal fluid, obtained through a lumbar puncture, can identify markers associated with CJD. The presence of 14-3-3 proteins in the spinal fluid is considered a significant indicator of the disease.
- Electroencephalogram (EEG): An EEG, which measures the electrical activity of the brain, can show characteristic patterns that suggest CJD. Although not specific to CJD alone, these patterns can support the diagnosis when combined with other test results.
These diagnostic tools, when used together, can significantly increase the accuracy of CJD diagnosis. However, it is important to note that a definitive diagnosis of CJD can only be made through a brain biopsy or an examination of brain tissue after death.
The Importance of Early Detection
Early detection of CJD is critical for several reasons. Firstly, it allows for the timely management of symptoms and the provision of supportive care, which can improve the quality of life for the patient and their family. Secondly, early detection is essential for the accurate classification of CJD type, which can have implications for public health and understanding the disease’s transmission. Lastly, identifying CJD early can help in the ongoing research and development of potential treatments and interventions.
However, the diagnosis and detection of Creutzfeldt-Jakob Disease hinge on a combination of clinical assessment, diagnostic tests, and an understanding of the disease’s progression. While there is currently no cure for CJD, early detection and accurate diagnosis are key to managing the disease’s symptoms and supporting affected individuals and their families. As research continues, it is hoped that more effective treatments will be discovered.
Treatment and Management of Creutzfeldt-Jakob Disease (CJD)
Despite the severity of CJD, current treatment options are limited, with an emphasis on providing supportive care to manage symptoms and improve the quality of life for those affected. Research into future treatments offers hope for advancements in managing or curing this devastating disease.
Current Treatment Options
As of now, there are no treatments available that can cure Creutzfeldt-Jakob disease or halt its progression. Medical intervention focuses on alleviating symptoms and making the patient as comfortable as possible. This symptomatic treatment may include:
- Medications: While no drugs can prevent the disease’s progression, certain medications can help manage symptoms such as muscle spasms, agitation, and pain.
- Safety Measures: Due to the loss of coordination and neurological function, patients may need adjustments in their living environment to prevent falls and injuries.
Supportive Care Strategies
Supportive care is central to the management of CJD, tailored to meet the individual needs of the patient and their family. This care approach encompasses a wide range of strategies, including:
- Physical Therapy: Aims to maintain muscle strength and mobility for as long as possible.
- Occupational Therapy: Helps adapt living spaces to ensure safety and promote independence.
- Nutritional Support: As swallowing difficulties may arise, dietary adjustments and feeding assistance might be necessary.
- Palliative Care: Focuses on relieving symptoms and improving the quality of life for both patients and their families. This may involve pain management, spiritual support, and counseling services.
Research and Future Treatments
The scientific community continues to investigate the causes and potential treatments for Creutzfeldt-Jakob disease. Some promising areas of research include:
- Antiprion Compounds: Researchers are exploring drugs that can target and inhibit the abnormal prion proteins responsible for CJD.
- Immunotherapy: This approach aims to enhance the immune system’s ability to fight off the harmful prion proteins.
- Gene Therapy: Early-stage research is looking into ways to correct or compensate for the genetic mutations that can cause inherited forms of CJD.
Although these treatments are still in the research phase, they represent a beacon of hope for those affected by Creutzfeldt-Jakob disease. Advances in medical science and technology continue to open new avenues for potentially effective treatments in the future.
However, while the current treatment landscape for Creutzfeldt-Jakob disease is primarily supportive, ongoing research into the mechanisms of the disease and potential therapeutic approaches holds promise for future advancements. By focusing on symptom management and supportive care, medical professionals strive to enhance the quality of life for individuals with CJD and their families.
Prevention Strategies for Creutzfeldt-Jakob Disease (CJD)
Understanding and implementing prevention strategies can significantly reduce the risk of transmission and occurrence. This guide outlines effective prevention strategies for CJD, focusing on guidelines for medical procedures, food safety measures, and genetic counseling for hereditary CJD.
Guidelines for Medical Procedures
- Sterilization of Surgical Instruments: Since CJD can be transmitted through contaminated medical equipment, it’s crucial that all surgical instruments undergo rigorous sterilization processes. The use of single-use instruments, when possible, is highly recommended.
- Use of Disposable Supplies: To minimize the risk of transmission, healthcare facilities should use disposable items whenever possible. This includes needles, scalpels, and other instruments that come into contact with high-infection-risk tissues.
- Tissue Handling Protocols: Healthcare workers should follow strict protocols when handling tissues and fluids from the brain or nervous system. This includes wearing protective gear and following specific procedures designed to prevent exposure.
Food Safety Measures
- Avoiding At-Risk Foods: While Variant Creutzfeldt-Jakob Disease (vCJD) is linked to consuming beef products contaminated with the Bovine Spongiform Encephalopathy (BSE) agent, adhering to food safety guidelines and avoiding at-risk foods can reduce transmission risk.
- Regulations on Animal Feed: Ensuring that cattle and other animals do not consume feed containing animal byproducts is key to preventing BSE, which in turn reduces the risk of vCJD in humans.
- Food Inspection and Control: Rigorous inspection and control measures in slaughterhouses and meat processing plants are essential to detect and remove potentially BSE-infected tissues from the food chain.
Genetic Counseling for Hereditary CJD
- Family History Assessment: Individuals with a family history of hereditary CJD should consider genetic counseling to understand their risk and discuss potential preventive measures.
- Genetic Testing: Genetic testing can identify mutations associated with hereditary CJD, offering an opportunity for early detection and planning for affected families.
- Support and Information: Genetic counselors provide valuable support and information to individuals at risk for hereditary CJD, including discussing reproductive options and strategies to manage or reduce the risk of transmission to future generations.
By adhering to these prevention strategies, individuals and healthcare providers can play a crucial role in reducing the risk of CJD transmission and occurrence. While CJD remains a challenging disease, proactive measures in medical procedures, food safety, and genetic counseling can significantly impact public health and safety.
Living with Creutzfeldt-Jakob Disease
Living with Creutzfeldt-Jakob Disease (CJD) presents unique challenges for both patients and their families. Navigating the journey requires understanding, patience, and a well-thought-out strategy to manage the symptoms and maintain quality of life. Below, we outline key aspects of coping with CJD, focusing on practical coping strategies, palliative care considerations, and available resources and support networks to assist patients and their families.
Coping Strategies for Patients and Families
Coping with a diagnosis of Creutzfeldt-Jakob Disease demands a comprehensive approach that addresses physical, emotional, and logistical challenges:
- Education: Understanding the disease can empower patients and families, helping them to make informed decisions about care and treatment. Knowledge about the progression of CJD and what to expect can reduce anxiety and help in planning for the future.
- Emotional Support: Emotional and psychological support is crucial. This can come from counseling, support groups, or therapy sessions aimed at helping patients and their loved ones deal with the emotional toll of the disease.
- Daily Routine Adjustments: Adjusting daily routines to accommodate the changing needs of the patient can help in managing symptoms more effectively. This may include modifications to the home environment to ensure safety and comfort.
- Legal and Financial Planning: Early planning for legal and financial matters, including wills, power of attorney, and healthcare directives, can alleviate future stresses and ensure the patient’s wishes are honored.
Palliative Care Considerations
Palliative care plays a vital role in the management of Creutzfeldt-Jakob Disease, focusing on relieving symptoms and improving the quality of life for both the patient and their family:
- Symptom Management: Palliative care teams specialize in managing symptoms such as pain, sleep disturbances, and mood swings, providing relief and comfort to patients.
- Holistic Support: Beyond physical symptoms, palliative care also addresses emotional, social, and spiritual needs, ensuring a comprehensive support system is in place.
- End-of-Life Care: As CJD progresses, palliative care becomes integral in facilitating end-of-life discussions and decisions, ensuring the patient’s comfort and dignity are maintained.
Resources and Support Networks
Finding the right resources and support networks is crucial for navigating the challenges of CJD:
- National and Local Support Groups: Organizations such as the Creutzfeldt-Jakob Disease Foundation offer resources, support groups, and hotlines to connect families with information and others going through similar experiences.
- Online Communities: Online forums and social media groups can provide a platform for sharing experiences, advice, and emotional support.
- Healthcare Team: Building a strong relationship with your healthcare team, including neurologists, palliative care specialists, and nurses, can provide guidance and support tailored to the patient’s needs.
With the right coping strategies, palliative care considerations, and access to supportive resources, it is possible to navigate these challenges more effectively. Empowerment through knowledge and support can make a significant difference in managing this disease.
Frequently Asked Questions (FAQs) About Creutzfeldt-Jakob Disease
What is Creutzfeldt-Jakob Disease (CJD)?
Creutzfeldt-Jakob Disease (CJD) is a rare, degenerative, invariably fatal brain disorder that affects about one in every one million people worldwide each year. CJD belongs to a family of human and animal diseases known as the transmissible spongiform encephalopathies (TSEs) due to the sponge-like holes that form in the brain. It progresses rapidly, leading to severe disability and death within months after symptoms appear.
How is CJD transmitted?
Most cases of CJD occur sporadically, meaning they arise in people with no known risk factors. However, it can also be inherited or acquired through exposure to brain or nervous system tissue, usually through certain medical procedures. There is no evidence that CJD is contagious through casual contact with a CJD patient or their bodily fluids.
What are the symptoms of CJD?
Symptoms of CJD can be similar to other progressive neurological disorders and may include rapidly worsening dementia, muscle stiffness, twitching, and weakness, difficulty walking, and changes in mood or behavior. As the disease progresses, mental impairment becomes severe, and involuntary movements, blindness, weakness of extremities, and coma may occur.
Is there a cure for CJD?
Currently, there is no cure for CJD. Treatment is focused on relieving symptoms and making the patient as comfortable as possible. This may include medications to manage pain, seizures, or agitation, and interventions to help with mobility, nutrition, and communication as the disease progresses.
How can I reduce my risk of getting CJD?
The risk of acquiring CJD is extremely low. To reduce any potential risk, avoid consuming brain or nervous system tissues from animals, especially in regions where bovine spongiform encephalopathy (BSE), also known as “mad cow disease,” has been found. In medical settings, strict procedures for sterilizing instruments and using synthetic growth hormone can prevent iatrogenic (medically acquired) cases.
Can CJD be inherited?
Yes, a small percentage of CJD cases (about 10-15%) are inherited. These cases are caused by a mutation in the PRNP gene, which can be passed from parent to child. Genetic testing is available for families with a history of CJD to determine if they carry the mutation.
How is CJD diagnosed?
Diagnosing CJD can be challenging and often involves ruling out other causes of dementia and neurological symptoms. Tests may include an electroencephalogram (EEG), magnetic resonance imaging (MRI) scans of the brain, and cerebrospinal fluid tests for specific proteins associated with CJD. In some cases, a brain biopsy may be necessary to confirm the diagnosis.
Conclusion:
We encourage readers to continue learning about Creutzfeldt-Jakob Disease and the broader category of prion diseases. Engaging with the latest research, participating in awareness campaigns, and supporting organizations dedicated to studying these diseases can all contribute to the global effort against CJD. The journey toward understanding and eventually overcoming Creutzfeldt-Jakob Disease is a challenging one, but with continued research and increased awareness, we can move closer to a future where this disease no longer poses the threat it currently does.
In conclusion, while Creutzfeldt-Jakob Disease remains a devastating condition with no current cure, the collective efforts of the medical community, researchers, and the public in raising awareness, conducting research, and supporting affected individuals and their families are invaluable. It is through these efforts that we can hope to see advancements in the understanding, management, and eventual eradication of CJD.


